La maladie de Caroli fait partie de ces pathologies hépatiques rares qui bouleversent le quotidien sans être connues du grand public. Lorsqu’un foie présente des dilatations kystiques des voies biliaires intra-hépatiques, les risques d’angiocholite, de calculs biliaires et de complications graves comme le cholangiocarcinome augmentent considérablement. Pour vous, patient, parent ou professionnel de santé, comprendre cette maladie congénitale, ses symptômes et les modalités de suivi médical à long terme est essentiel pour anticiper les complications et améliorer la qualité de vie. Une prise en charge coordonnée au sein de centres experts des maladies rares du foie, comme ceux du réseau français FilFoie, change radicalement le pronostic, surtout lorsque le diagnostic est posé tôt et que le suivi est structuré.
Définition de la maladie de caroli : distinction entre forme simple et syndrome de caroli (Caroli-Meyenburg)
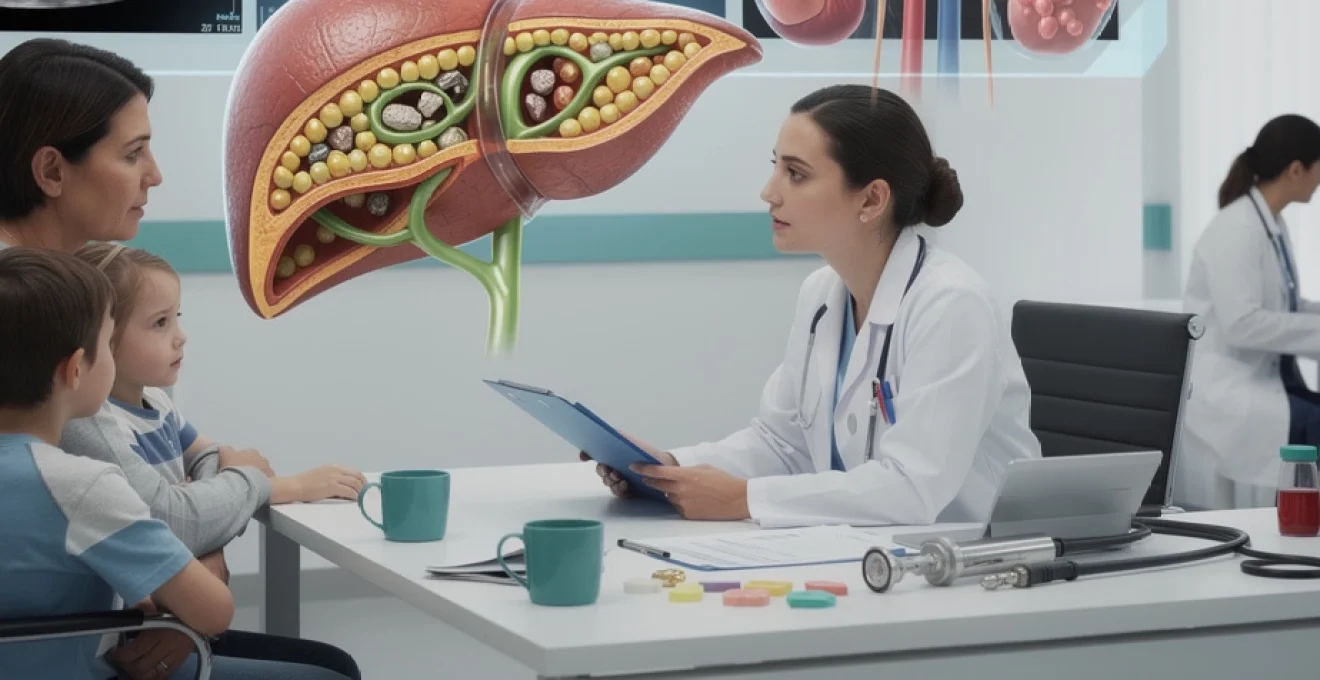
La maladie de Caroli est une affection hépatobiliaire congénitale rare, caractérisée par une dilatation kystique segmentaire non obstructive des voies biliaires intra-hépatiques de gros calibre. Ces dilatations donnent à l’imagerie un aspect en “chapelet” très évocateur. Dans la forme dite simple, il s’agit essentiellement d’une ectasie biliaire sans autre anomalie majeure du parenchyme hépatique. Le foie garde une architecture relativement conservée, même si des épisodes d’infection ou de lithiase peuvent l’altérer progressivement.
Le syndrome de Caroli (souvent appelé Caroli-Meyenburg) correspond à la forme syndromique, plus fréquente, où la dilatation des canaux biliaires intra-hépatiques s’associe à une fibrose hépatique congénitale. Cette fibrose peut évoluer vers une véritable cirrhose biliaire avec hypertension portale, splénomégalie et varices œsophagiennes. Dans la pratique, de nombreux patients classés “Caroli” présentent en réalité ce spectre syndromique. Les données d’Orphanet confirment que le syndrome de Caroli est plus courant que la forme strictement isolée.
Physiopathologie de la dilatation des voies biliaires intra-hépatiques segmentaires
Au cœur de la physiopathologie de la maladie de Caroli se trouve une anomalie de développement de la plaque ductale, structure embryonnaire à partir de laquelle se forment les voies biliaires intrahépatiques. Normalement, cette plaque se remodèle pour donner un réseau ordonné de canaux. En cas de malformation, certaines portions restent dilatées, kystiques, et communiquent avec l’arbre biliaire principal. Ces “poches” de bile stagnante favorisent la prolifération bactérienne, la formation de boue biliaire et de calculs pigmentaires.
Cette stagnation chronique agit un peu comme une rivière ralentie par des méandres trop larges : les dépôts s’accumulent, l’écoulement devient irrégulier, et les berges (ici la paroi biliaire) se fragilisent. Sur le long terme, cette architecture anormale du réseau biliaire entraîne des épisodes répétés de cholangite, source d’inflammation chronique et de remodelage fibreux du foie, surtout dans le contexte du syndrome de Caroli.
Différences entre maladie de caroli isolée et fibrose hépatique congénitale associée
Dans la maladie de Caroli isolée, les lésions se limitent en principe aux voies biliaires intra-hépatiques. Le parenchyme hépatique reste peu ou pas fibrosé en dehors des zones adjacentes aux dilatations. Les manifestations dominantes sont alors les lithiases biliaires intrahépatiques, les épisodes de colique hépatique, les angiocholites récidivantes et, plus rarement, une pancréatite aiguë.
Dans le syndrome de Caroli, la fibrose hépatique congénitale diffuse modifie complètement le profil clinique. Les patients présentent plus souvent une hypertension portale précoce, avec splénomégalie, hypersplénisme, thrombopénie, et risque d’hémorragies digestives par rupture de varices œsophagiennes. Le foie peut évoluer vers une cirrhose biliaire secondaire, même en l’absence de grosses cholangites. Cette distinction est capitale pour estimer le risque d’insuffisance hépatique et discuter précocement une transplantation.
Classification OMS des cholangio-pathies congénitales (caroli, von meyenburg, kystes biliaires)
Sur le plan anatomo-pathologique, la maladie de Caroli s’intègre dans le groupe des malformations fibrokystiques hépatiques. La classification internationale distingue :
- La maladie de Caroli et le syndrome de Caroli (dilatations biliaires de gros calibre)
- Les complexes de
Von Meyenburg(microhamartomes biliaires multiples, souvent découverts fortuitement) - Les kystes biliaires isolés et la polykystose hépatique
Cette classification aide à faire la part entre lésions purement kystiques, hamartomateuses et dilatations communicantes avec l’arbre biliaire. Dans la pratique clinique, la distinction la plus utile reste celle entre maladie de Caroli simple, syndrome de Caroli et autres kystes hépatiques non communicants, car les implications pronostiques et thérapeutiques sont très différentes.
Épidémiologie : maladie rare, prévalence, cas familiaux et formes sporadiques
La maladie de Caroli est considérée comme une maladie ultra-rare. Les estimations parlent d’une prévalence d’environ 1 cas pour 1 000 000 habitants, avec quelques centaines de cas décrits dans la littérature internationale. Certaines séries suggèrent une légère prédominance féminine et un âge moyen de diagnostic autour de 30–40 ans chez l’adulte, même si plus de 80 % des cas débuteraient avant 30 ans.
La plupart des formes isolées semblent spontanées ou sporadiques, alors que le syndrome de Caroli est souvent retrouvé dans un contexte familial, associé à une polykystose rénale autosomique récessive. Dans certains pays à forte consanguinité, le nombre de cas familiaux est plus élevé. L’amélioration des techniques d’imagerie a conduit depuis une dizaine d’années à une légère hausse des diagnostics, y compris en période néonatale grâce à l’échographie prénatale, mais sans explosion de l’incidence.
Étiologie génétique et mécanismes moléculaires impliqués dans la maladie de caroli
L’étiologie de la maladie de Caroli simple reste encore partiellement débattue, avec des cas décrits comme sporadiques. En revanche, les formes syndromiques et familiales sont fortement liées à des mutations du gène PKHD1 (Polycystic Kidney and Hepatic Disease 1). Ce gène code pour la protéine fibrocystine/polyductine, essentielle au développement harmonieux des tubules rénaux et des voies biliaires. Les ressources spécialisées comme Maladie de Caroli – Filfoie et Maladie de Caroli – Orphanet détaillent ce lien génétique et son impact sur la prise en charge.
Mutations du gène PKHD1 (fibrocystine/polyductine) et transmission autosomique récessive
Le gène PKHD1, localisé sur le chromosome 6p, suit un mode de transmission autosomique récessive. Pour qu’un enfant développe un syndrome de Caroli lié à PKHD1, il doit hériter de deux copies mutées, l’une provenant de chaque parent porteur sain. Le risque de transmission à chaque grossesse est donc de 25 % si les deux parents sont hétérozygotes.
Les mutations de PKHD1 altèrent la structure de la fibrocystine, impliquée dans la polarité cellulaire et l’organisation des cils primaires des cholangiocytes. Ces cils jouent un rôle de “capteurs” mécaniques et chimiques. Lorsqu’ils dysfonctionnent, l’architecture tubulaire des canaux biliaires se dérègle, donnant lieu à des zones de dilatation kystique communiquant avec le réseau biliaire principal, typiques de la maladie de Caroli.
Lien entre maladie de caroli et polykystose rénale autosomique récessive (ARPKD)
La polykystose rénale autosomique récessive (ARPKD) partage le même gène PKHD1 que le syndrome de Caroli. De nombreux patients atteints d’ARPKD présentent aussi des anomalies biliaires et une fibrose hépatique congénitale, d’où l’association fréquente Caroli–ARPKD. Dans ces formes, les reins sont volumineux, dysplasiques, avec une insuffisance rénale chronique parfois sévère dès l’enfance.
Sur le plan clinique, cette association se traduit par une double atteinte : hépato-biliaire et rénale. Vous pouvez alors observer chez un même patient hématurie, hypertension artérielle, baisse du débit de filtration glomérulaire, en plus des signes de cholestase et d’hypertension portale. Les travaux récents présentés dans plusieurs congrès de néphrologie pédiatrique insistent sur la nécessité d’un dépistage systématique des anomalies biliaires chez les enfants porteurs d’ARPKD.
Anomalies du développement des plaques ductales (ductal plate malformation)
Le concept de ductal plate malformation désigne un défaut de remodelage de la plaque ductale au cours de l’embryogenèse. Plutôt qu’un processus linéaire, il s’agit d’un spectre d’anomalies, depuis les complexes de Von Meyenburg jusqu’à la polykystose hépatique. La maladie de Caroli et le syndrome de Caroli se situent dans cette continuité.
Une façon simple d’imaginer cette malformation est de penser à un réseau de canalisations initialement circulaires autour des branches portales qui doivent se remodeler en tubes fins et organisés. Si ce remodelage reste incomplet, certaines portions deviennent des “culs-de-sac” dilatés où la bile tourne en rond. Ces anomalies de développement sont modulées par les gènes impliqués dans la ciliogénèse et le maintien de la matrice extracellulaire, ce qui explique les nombreux liens avec d’autres maladies ciliopathiques.
Exploration génétique : séquençage ciblé, panels NGS et conseil génétique familial
Sur le plan pratique, l’exploration génétique repose aujourd’hui sur des techniques de séquençage de nouvelle génération (NGS). Dans les formes syndromiques ou familiales, un panel ciblé incluant PKHD1 et d’autres gènes de maladies fibrokystiques rénales et hépatiques est proposé. Le séquençage complet de PKHD1 reste complexe en raison de la taille du gène et du nombre de variants, mais les plateformes spécialisées y parviennent de plus en plus rapidement.
Le conseil génétique familial occupe une place majeure : pour vous, parent concerné, il permet de comprendre le risque de récurrence, de discuter d’un éventuel diagnostic prénatal ou pré-implantatoire, et d’orienter le dépistage des frères et sœurs. Les recommandations de réseaux comme Syndrome de Caroli – Filfoie et de bases de données comme Orphanet insistent sur cette approche intégrée.
Symptômes cliniques typiques de la maladie de caroli chez l’adulte et l’enfant
Les manifestations cliniques de la maladie de Caroli sont très variables. Certains patients restent asymptomatiques pendant des années, avec une découverte fortuite à l’imagerie, tandis que d’autres, enfants ou jeunes adultes, enchaînent les hospitalisations pour cholangites récidivantes. Les symptômes sont essentiellement liés à la stase biliaire, à l’infection et, dans le syndrome de Caroli, à la fibrose hépatique et à l’atteinte rénale associée.
Douleurs de l’hypochondre droit, fièvre et ictère liés aux épisodes de cholangite
Le tableau classique de cholangite aiguë associe :
- Douleur abdominale de l’hypochondre droit ou épigastrique
- Fièvre souvent supérieure à 38,5 °C avec frissons
- Ictère cutanéo-muqueux plus ou moins intense
Cette triade, parfois complétée par une altération de l’état général, doit alerter sur une infection biliaire grave. Dans la maladie de Caroli, ces épisodes sont favorisés par les dilatations segmentaires qui retiennent la bile et les bactéries. Une angiocholite non traitée peut évoluer vers un sepsis sévère, justifiant une hospitalisation urgente, une antibiothérapie intraveineuse et parfois un drainage biliaire.
Lithiases biliaires intra-hépatiques, prurit et épisodes de cholestase prolongée
Plus de 90 % des patients présentent, au cours de l’évolution, des calculs intra-hépatiques ou une micro-lithiase biliaire. Ces calculs obstruent les canaux dilatés, aggravent la stase et provoquent des épisodes de cholestase prolongée. Pour vous, cela peut se traduire par :
Un prurit souvent diffuse, parfois insomniant, des urines foncées et des selles décolorées. Les tests hépatiques montrent alors une élévation des phosphatases alcalines, de la GGT et de la bilirubine. Ces poussées cholestatiques répétées participent à la dégradation progressive du parenchyme hépatique et augmentent, à long terme, le risque de transformation maligne.
Manifestations extra-hépatiques : hématurie, insuffisance rénale, HTA dans les formes associées ARPKD
Dans les formes associées à une polykystose rénale autosomique récessive, vous pouvez observer des signes extra-hépatiques marqués : hématurie macroscopique ou microscopique, infections urinaires, douleurs lombaires, mais aussi une hypertension artérielle précoce et une insuffisance rénale progressive. Les reins sont souvent augmentés de taille, bosselés, avec de multiples microkystes corticaux.
Ces manifestations imposent un suivi conjoint par un néphrologue et un hépatologue. Les données des registres pédiatriques montrent qu’environ 30 à 50 % des enfants atteints d’ARPKD développent aussi une atteinte biliaire de type Caroli ou fibrose hépatique, avec un risque significatif d’hypertension portale et de cholangite dès l’adolescence.
Signes de maladie hépatique chronique : splénomégalie, varices œsophagiennes et ascite
Dans le syndrome de Caroli, la fibrose hépatique congénitale peut se comporter comme une cirrhose biliaire. Les signes d’hypertension portale sont fréquents : splénomégalie parfois massive, thrombopénie, varices œsophagiennes visibles à la fibroscopie, et, plus tardivement, ascite. Vous pouvez remarquer une augmentation progressive du volume de l’abdomen, des hématomes cutanés plus fréquents ou des épisodes d’hématémèse.
Les hémorragies digestives par rupture de varices œsophagiennes représentent l’une des complications les plus redoutées du syndrome de Caroli, justifiant une surveillance endoscopique proactive.
La prise en charge de cette dimension “cirrhotique” suit les mêmes principes que pour les autres causes d’hypertension portale, avec bêtabloquants non sélectifs, ligatures endoscopiques et, dans certains cas, mise en place d’un TIPS (shunt porto-systémique intrahépatique par voie transjugulaire).
Diagnostic de la maladie de caroli : imagerie hépatobiliaire et examens complémentaires
Le diagnostic repose avant tout sur l’imagerie hépatobiliaire. Les progrès de l’échographie, du scanner et surtout de la cholangio-IRM ont considérablement affiné la détection et la caractérisation des dilatations biliaires. Les recommandations disponibles sur des sites spécialisés comme Syndrome de Caroli – Filfoie et les fiches explicatives des centres universitaires d’hépato-gastroentérologie permettent de standardiser ce bilan.
Échographie abdominale avec visualisation des dilatations kystiques segmentaires
L’échographie abdominale constitue souvent le premier examen réalisé devant une douleur de l’hypochondre droit, un ictère ou une hépatomégalie. Elle montre des images anéchogènes ou hypoéchogènes intrahépatiques, arrondies, correspondant aux dilatations kystiques des canaux biliaires. Ces cavités communiquent parfois entre elles, mais l’échographie seule ne permet pas toujours de le confirmer.
Chez l’enfant, l’échographie est particulièrement utile, car non irradiant et facilement répétable. Certains cas de maladie de Caroli et de syndrome de Caroli ont même été identifiés en période prénatale, au troisième trimestre, devant des images kystiques hépatiques multiples. Dans ces situations, un bilan postnatal complet est indispensable pour préciser la nature des lésions.
IRM cholangio-pancréatique (Cholangio-IRM) : signe du « central dot sign » et cartographie des voies biliaires
La cholangio-IRM est l’examen de référence pour la maladie de Caroli. Grâce à des séquences fortement pondérées en T2, elle met en évidence les dilatations kystiques communicant avec l’arbre biliaire. Le signe caractéristique est le central dot sign : un point hyperintense correspondant à une branche portale entourée par la cavité biliaire dilatée.
Cette technique permet une véritable “cartographie” des voies biliaires intra-hépatiques, en distinguant les atteintes unilobaires des formes diffuses. Elle aide aussi à différencier la maladie de Caroli d’autres pathologies comme la cholangite sclérosante primitive ou les kystes hépatiques simples. Plusieurs études d’imagerie publiées ces dernières années montrent une sensibilité diagnostique supérieure à 90 % lorsque la cholangio-IRM est correctement réalisée et interprétée.
Scanner multiphases du foie : bilan d’extension, recherche de cholangiocarcinome et calculs biliaires
Le scanner hélicoïdal multiphases complète l’IRM en apportant une vision fine du parenchyme hépatique, des vaisseaux et des éventuelles lésions tumorales. Il permet de rechercher un cholangiocarcinome intra-hépatique, complication maligne décrite dans 5 à 10 % des cas selon certaines séries, et de détecter des calculs intra-hépatiques radio-opaques.
Le scanner est également utile pour planifier une éventuelle résection hépatique ou une transplantation, en évaluant la répartition des lésions, le volume de foie sain et la perméabilité du système porte. Les sociétés savantes recommandent souvent un couplage IRM + scanner dans les bilans pré-chirurgicaux complexes.
Cholangiographie rétrograde endoscopique (CPRE) et cholangioscopie pour bilan des sténoses et lithiases
La CPRE (cholangiopancréatographie rétrograde endoscopique) n’est plus recommandée pour le diagnostic initial en raison de son caractère invasif et du risque de déclencher une cholangite. Elle conserve cependant une place de choix en tant que geste thérapeutique : extraction de calculs, dilatation de sténoses, mise en place de prothèses biliaires.
Dans certains centres, la cholangioscopie par voie endoscopique permet une exploration directe de la lumière des canaux biliaires, utile pour affiner le bilan des lésions et prélever des biopsies ciblées si une transformation maligne est suspectée. Ces techniques interventionnelles doivent être réservées à des équipes expérimentées, souvent dans des centres experts des maladies biliaires rares.
Bilan biologique hépatique : transaminases, phosphatases alcalines, GGT, bilirubine et marqueurs tumoraux (CA 19-9)
Le bilan biologique hépatique inclut systématiquement : ALAT, ASAT, phosphatases alcalines, GGT, bilirubine totale et conjuguée, albumine et temps de prothrombine. En période de cholangite, la CRP et la numération leucocytaire sont élevées. Sur le long terme, une hypoalbuminémie et un allongement du TP signent une insuffisance hépatocellulaire.
Le dosage du CA 19‑9 est souvent intégré au suivi, bien qu’il manque de spécificité. Une élévation progressive et persistante doit inciter à rechercher un cholangiocarcinome, surtout en cas de modification morphologique suspecte à l’imagerie. Les recommandations des centres universitaires et des réseaux de référence, accessibles via des portails comme Orphanet, soulignent l’importance de la corrélation clinique et radiologique.
Complications de la maladie de caroli : cholangites récidivantes, lithiases et cholangiocarcinome
Les complications conditionnent le pronostic et la qualité de vie. Sans surveillance ni prise en charge adaptée, la maladie de Caroli peut évoluer vers des cholangites bactériennes à répétition, une lithiase biliaire complexe, une hypertension portale et, plus rarement, un cholangiocarcinome intra-hépatique. Pour vous, l’enjeu est de reconnaître tôt ces complications afin d’adapter les stratégies thérapeutiques.
Cholangites bactériennes aiguës à répétition : germes fréquents, sepsis et hospitalisations
Les cholangites bactériennes sont essentiellement dues à des germes entériques : Escherichia coli, Klebsiella, entérocoques, parfois anaérobies. La stagnation de la bile dans les canaux dilatés favorise leur ascension depuis le duodénum. Chaque épisode impose une antibiothérapie probabiliste intraveineuse, ajustée ensuite sur l’antibiogramme.
Les études cliniques estiment que 60 à 80 % des patients symptomatiques auront au moins un épisode de cholangite au cours de leur vie, et une proportion significative en présentera plusieurs par an. Ces épisodes sont l’une des principales causes d’hospitalisation et altèrent fortement la qualité de vie, sans compter le risque de sepsis sévère, notamment chez les patients immunodéprimés ou cirrhotiques.
Formation de calculs biliaires intra-hépatiques et sténoses segmentaires des canaux biliaires
Les calculs intra-hépatiques, souvent pigmentaires ou mixtes, se développent sur la bile épaissie et infectée. Ils peuvent occuper des poches kystiques entières, obstruer des canaux segmentaires et constituer des foyers d’infection chronique. À long terme, cette lithiase favorise la fibrose péri-canalaire et la formation de sténoses complexes.
Ces sténoses sont parfois accessibles à la dilatation endoscopique ou radiologique, mais dans d’autres cas, une résection hépatique segmentaire ciblée reste la meilleure option, surtout si l’atteinte est unilatérale. Plusieurs séries chirurgicales récentes rapportent une diminution majeure des épisodes de cholangite et une amélioration de la qualité de vie après hépatectomie partielle bien indiquée.
Risque de transformation maligne : cholangiocarcinome intra-hépatique et protocole de surveillance
La maladie de Caroli est un facteur de risque reconnu de cholangiocarcinome intra-hépatique. Le risque cumulé est estimé entre 5 et 10 % dans certaines cohortes, avec un risque multiplié par 100 par rapport à la population générale. Cette transformation survient classiquement après plusieurs années de maladie, sur un terrain de cholangite chronique et de lithiase.
La surveillance oncologique structurée, associant imagerie régulière et biomarqueurs, est l’un des piliers du suivi à long terme des patients atteints de maladie de Caroli.
Un protocole de surveillance typique comprend une échographie ou une IRM hépatique annuelle, complétée par un scanner en cas d’anomalie suspecte, ainsi qu’un dosage sériel du CA 19‑9. En présence d’une lésion nodulaire nouvelle ou d’une variation de signal atypique, une évaluation en RCP (réunion de concertation pluridisciplinaire) spécialisée est recommandée pour discuter biopsie, résection locale ou transplantation.
Hypertension portale et progression vers cirrhose biliaire secondaire
Dans le syndrome de Caroli, l’association fibrose hépatique congénitale + cholangites répétées peut conduire à une cirrhose biliaire secondaire. L’hypertension portale se manifeste alors par les signes décrits plus haut : splénomégalie, varices œsophagiennes, ascite, encéphalopathie hépatique dans les formes avancées.
Les données disponibles suggèrent que 20 à 40 % des patients présentant un syndrome de Caroli sévère développeront, au cours de leur vie, une hypertension portale significative. Cette évolution impose d’intégrer très tôt les outils classiques de prévention des hémorragies digestives (bêtabloquants, ligatures) et de discuter à temps l’option de la transplantation hépatique totale en cas de décompensations répétées.
Prise en charge thérapeutique de la maladie de caroli : stratégies médicales, endoscopiques et chirurgicales
La prise en charge de la maladie de Caroli repose sur un arsenal thérapeutique multimodal. Il n’existe pas, à ce jour, de traitement curatif médical de la malformation elle-même, mais une combinaison d’antibiothérapie, d’endoscopie interventionnelle, de chirurgie ciblée et, pour certains, de transplantation hépatique permet de contrôler les symptômes et de réduire le risque de complications. Les approches décrites dans les ressources spécialisées comme Maladie de Caroli – Filfoie et dans les centres de transplantation hépatique illustrent cette stratégie graduée.
Traitement médical des cholangites : antibiothérapie probabiliste, drainage biliaire et prévention des récidives
Lors d’une cholangite aiguë, la priorité est de stabiliser le patient : réhydratation, antibiothérapie probabiliste à large spectre (souvent céphalosporines de 3e génération ou fluoroquinolones associées à un imidazolé), puis adaptation aux résultats des hémocultures. En cas de choc septique, une prise en charge en soins intensifs est parfois nécessaire.
La prévention des récidives repose sur le contrôle de la stase biliaire : extraction des calculs, amélioration du drainage par endoscopie ou radiologie interventionnelle, parfois prophylaxie antibiotique chez les patients présentant des épisodes très fréquents. Les recommandations récentes insistent aussi sur la vaccination contre l’hépatite A et B, et sur l’optimisation de l’état nutritionnel pour diminuer la vulnérabilité aux infections.
Endoscopie interventionnelle (CPRE) : extraction de calculs, sphinctérotomie et dilatation de sténoses
La CPRE est l’outil de choix pour traiter les calculs situés dans les voies biliaires extra-hépatiques ou les branches proximales accessibles. La sphinctérotomie endoscopique facilite l’extraction de calculs, et la mise en place de stents plastiques ou métalliques permet de maintenir un bon drainage dans les cas de sténoses serrées.
Dans la maladie de Caroli, l’endoscopie ne permet pas toujours d’atteindre les segments intra-hépatiques distaux, mais elle constitue souvent une étape indispensable pour limiter la charge lithiasique globale et réduire la pression sur l’arbre biliaire. L’expérience des centres d’endoscopie avancée montre que des sessions répétées peuvent, chez certains patients bien sélectionnés, éviter ou retarder le recours à une chirurgie lourde.
Chirurgie hépatobiliaire ciblée : hépatectomie segmentaire ou lobaire dans les atteintes unilatérales
Lorsque les lésions de Caroli sont unilatérales (atteinte d’un seul lobe ou d’un seul segment), une hépatectomie segmentaire ou lobaire représente souvent le meilleur traitement à long terme. En retirant le territoire biliaire pathologique, cette chirurgie réduit drastiquement le risque de cholangite, de lithiase récidivante et de transformation maligne dans la zone concernée.
Les séries chirurgicales publiées rapportent des taux de survie globale à 10 ans supérieurs à 80 % pour ces formes unilobaires traitées de manière radicale. La sélection des candidats repose sur une évaluation volumétrique du foie sain, la fonction hépatique résiduelle et l’absence de contre-indication anesthésique majeure. Pour vous, cette option est à envisager précocement si l’imagerie montre une localité bien circonscrite des lésions.
Transplantation hépatique totale pour formes diffuses compliquées ou associées à fibrose hépatique congénitale
Dans les formes diffuses de maladie de Caroli, particulièrement lorsqu’elles s’associent à une fibrose hépatique congénitale sévère, l’option de la transplantation hépatique totale doit être envisagée. Les indications majeures incluent : cholangites récidivantes non contrôlables, hypertension portale compliquée, suspicion ou survenue de cholangiocarcinome résécable, et insuffisance hépatique avancée.
Les résultats à long terme de la transplantation dans ces indications sont encourageants, avec des taux de survie à 5 ans comparables à d’autres indications cholestatiques. Dans certains cas, une double transplantation foie–rein peut être discutée chez les patients présentant une polykystose rénale autosomique récessive terminale associée. Les centres de référence en transplantation hépato-rénale coordonnent alors un parcours extrêmement spécialisé.
Rôle de l’ursodésoxycholique et des traitements adjuvants dans la prise en charge au long cours
L’acide ursodésoxycholique (UDCA) est souvent prescrit au long cours dans la maladie de Caroli, avec l’objectif d’améliorer la fluidité de la bile, de réduire la formation de boue biliaire et de calculs, et d’atténuer le prurit. Bien qu’il ne corrige pas la malformation, plusieurs études montrent une diminution de la fréquence des poussées cholestatiques chez certains patients.
D’autres traitements adjuvants incluent les agents antiprurigineux (cholestyramine, rifampicine, parfois nalfurafine dans les essais récents), la supplémentation en vitamines liposolubles (A, D, E, K) en cas de cholestase chronique, et une prise en charge nutritionnelle personnalisée. Pour vous, une bonne adhésion à ces traitements de fond peut faire la différence sur le confort quotidien et la prévention des carences.
Suivi médical à long terme chez les patients atteints de maladie de caroli
Le suivi à long terme est primordial pour anticiper les complications infectieuses, métaboliques et oncologiques. Il repose sur un programme structuré de consultations spécialisées, d’examens d’imagerie et de bilans biologiques réguliers. Les recommandations issues des réseaux de référence français et européens, accessibles via des portails comme Orphanet, servent de base pour adapter ce suivi à chaque profil de patient.
Programme de consultations régulières en hépatologie : bilan clinique, biologique et imagerie annuelle
Un programme de suivi typique comprend :
| Fréquence | Éléments du suivi |
|---|---|
| Tous les 6 à 12 mois | Consultation d’hépatologie, bilan clinique, évaluation des symptômes (douleur, prurit, fatigue) |
| Au moins une fois par an | Bilan biologique complet (fonction hépatique, rénale, coagulation, CA 19‑9) |
| Selon le risque | Échographie ou IRM hépatique, fibroscopie digestive (si hypertension portale) |
Ce rythme est ajusté en fonction de la sévérité de la maladie, de la présence d’un syndrome de Caroli, de l’âge et des traitements en cours. En cas de transplantation, le suivi suit les protocoles habituels de post-greffe, avec un contrôle serré dans les premières années.
Surveillance ciblée du cholangiocarcinome : échographie, IRM hépatique et dosage sériel du CA 19-9
La surveillance du cholangiocarcinome repose sur la répétition régulière des examens d’imagerie et des marqueurs tumoraux. L’échographie annuelle est un minimum, souvent complétée par une IRM hépatique tous les 1 à 2 ans chez les patients à haut risque (formes diffuses, antécédents de cholangite fréquente, lithiase abondante).
Le dosage sériel du CA 19‑9, même imparfait, contribue à la détection précoce lorsqu’il est interprété dans le contexte. Une hausse brutale ou un niveau très élevé doit inciter à réaliser rapidement un scanner multiphasique et, si besoin, une biopsie ciblée. L’enjeu est de diagnostiquer les lésions à un stade potentiellement résécable ou éligible à une stratégie de transplantation curative.
Prise en charge multidisciplinaire : hépatologue, néphrologue, radiologue, chirurgien et généticien
La complexité de la maladie de Caroli impose une approche multidisciplinaire. Un même patient peut nécessiter l’expertise :
- D’un hépatologue ou hépato-gastroentérologue pour le suivi hépatique
- D’un néphrologue en cas d’ARPKD associée ou d’atteinte rénale
- D’un radiologue spécialisé en imagerie hépatobiliaire
- D’un chirurgien hépatobiliaire et d’une équipe de transplantation
Un généticien clinicien intervient pour le conseil familial et l’interprétation des variants de PKHD1. Les réunions de concertation pluridisciplinaire, telles que celles organisées dans les centres universitaires référencés par FilFoie, sont au cœur de cette organisation, afin de proposer des décisions personnalisées et argumentées.
Suivi spécifique chez l’enfant : croissance, développement pubertaire et adaptation des posologies
Chez l’enfant, la maladie de Caroli et le syndrome de Caroli soulèvent des enjeux spécifiques : impact sur la croissance staturo-pondérale, sur le développement pubertaire, sur la scolarité et la vie familiale. Les posologies des médicaments (UDCA, antibiotiques, antihypertenseurs) doivent être régulièrement ajustées en fonction du poids et de la maturation rénale et hépatique.
Les pédiatres hépatologues insistent également sur la nécessité d’un soutien psychologique et scolaire adapté. Les études récentes en médecine de l’adolescent montrent que les pathologies hépatiques rares, lorsqu’elles sont bien expliquées et intégrées dans un projet de vie, n’empêchent pas un parcours normal, y compris pour les patients transplantés.
Éducation thérapeutique du patient : prévention des infections biliaires, vaccination et hygiène de vie
L’éducation thérapeutique joue un rôle majeur pour vous aider à devenir acteur du suivi. Elle inclut des conseils pratiques pour reconnaître les signes de cholangite (fièvre, frissons, douleur, ictère), pour consulter sans délai en cas de symptômes d’alerte, et pour respecter les traitements au long cours.
Une bonne compréhension de la maladie par le patient et sa famille réduit significativement les retards de prise en charge lors des épisodes infectieux et améliore l’adhésion thérapeutique.
La vaccination contre les hépatites virales A et B, le pneumocoque et la grippe saisonnière est recommandée, surtout si un traitement immunosuppresseur est envisagé (post-transplantation). Une hygiène de vie adaptée — alimentation équilibrée, limitation de l’alcool, activité physique régulière — contribue à préserver le capital hépatique et rénal au long cours.
Pronostic, qualité de vie et ressources pour les patients atteints de maladie de caroli
Le pronostic de la maladie de Caroli varie considérablement selon la forme (simple ou syndromique), l’extension des lésions biliaires, la présence d’une polykystose rénale associée et l’accès à une prise en charge spécialisée. Les ressources disponibles via des réseaux dédiés aux maladies rares du foie, tels que FilFoie, offrent des informations fiables et orientent vers les centres experts les plus proches.
Facteurs pronostiques : extension des lésions biliaires, fréquence des cholangites et atteinte rénale associée
Les principaux facteurs pronostiques identifiés sont :
| Facteur | Impact sur le pronostic |
|---|---|
| Extension des lésions (unilatérale vs diffuse) | Formes unilatérales résécables : meilleur pronostic à long terme |
| Fréquence des cholangites | Plus les épisodes sont fréquents, plus le risque de fibrose et de cholangiocarcinome augmente |
| Atteinte rénale (ARPKD) | Insuffisance rénale chronique et hypertension artérielle aggravent la morbidité globale |
| Présence d’hypertension portale | Risque accru d’hémorragies digestives et de décompensation hépatique |
Les données de survie montrent que, dans la forme pure de la maladie de Caroli bien contrôlée, la survie à 10 ans dépasse 80 %, tandis que les formes syndromiques sévères associées à une fibrose et à une ARPKD présentent un pronostic plus réservé sans transplantation.
Impact sur la qualité de vie : douleurs chroniques, fatigue, contraintes de suivi hospitalier
Au-delà des chiffres, l’impact sur la qualité de vie est majeur. Douleurs abdominales chroniques, fatigue liée aux épisodes infectieux, prurit, hospitalisations répétées, examens invasifs ou lourds (endoscopies, interventions radiologiques, chirurgies) pèsent sur le quotidien. Pour vous, ces contraintes peuvent retentir sur la vie professionnelle, scolaire, sociale et familiale.
Les études de qualité de vie menées dans les maladies cholestatiques rares montrent une prévalence accrue de troubles anxieux et dépressifs. Un accompagnement psychologique, l’accès à des groupes de parole et à des associations de patients spécialisés dans les maladies du foie de l’enfant et de l’adulte constituent des ressources précieuses pour rompre l’isolement et partager des stratégies d’adaptation concrètes.
Espérance de vie et résultats à long terme après transplantation hépatique
Pour les patients ayant bénéficié d’une transplantation hépatique pour maladie ou syndrome de Caroli, les résultats à long terme sont globalement favorables. Les séries des grandes équipes montrent des taux de survie à 5 ans proches de 80–85 %, et une qualité de vie nettement améliorée, avec disparition des cholangites et du prurit, et contrôle de l’hypertension portale.
L’espérance de vie après transplantation dépend surtout des complications liées à l’immunosuppression, de la survenue éventuelle de rejet chronique et, en cas de greffe combinée foie–rein, de l’évolution de la fonction rénale. Les progrès des protocoles immunosuppresseurs, présentés chaque année dans les grands congrès de transplantation, tendent à réduire les effets indésirables et à améliorer la tolérance à long terme.
Réseaux de référence et centres experts en hépatologie rare en france (FilFoie, centres universitaires)
En France, la prise en charge de la maladie de Caroli et du syndrome de Caroli s’appuie sur des réseaux de référence comme FilFoie, qui coordonne les centres experts d’hépatologie rare pédiatrique et adulte. Ces centres, implantés au sein de CHU et d’hôpitaux universitaires, regroupent les compétences en hépatologie, néphrologie, radiologie interventionnelle, chirurgie hépatobiliaire et génétique.
Pour vous ou votre enfant, être suivi dans l’un de ces centres offre un accès facilité aux avis spécialisés, aux techniques d’imagerie avancée, aux interventions complexes et aux essais cliniques en cours, notamment sur les nouvelles approches d’immunothérapie et de thérapies ciblées dans les cancers des voies biliaires associés. Les bases de données comme Orphanet permettent d’identifier rapidement ces structures référentes et de mieux comprendre les ressources disponibles pour accompagner la maladie sur le long terme.